O priões são proteínas sem genoma ou ácidos nucléicos que atuam como agentes infecciosos. O termo "príon" significa partícula infecciosa proteica (do inglês Proteinaceous Infectious Particles), e foi cunhado pelo neurologista e vencedor do Prêmio Nobel, Stanley B. Prusiner.
Em 1982, Prusiner e seus colegas identificaram uma partícula de proteína infecciosa, enquanto estudavam as causas das doenças de Creutzfeldt-Jakob (em humanos) e da encefalopatia espongiforme bovina..
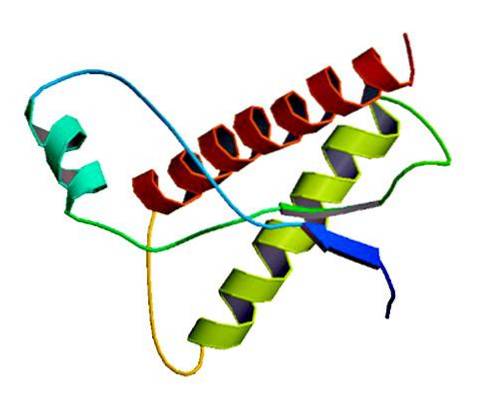
Esses raros agentes infecciosos são encontrados na membrana das células normais, apenas como proteínas mal dobradas e / ou com estrutura tridimensional anormal. Essas proteínas são responsáveis por múltiplas doenças degenerativas e altíssima mortalidade que afetam os tecidos neurais e a estrutura do cérebro..
Eles também são chamados de doenças de príons. Entre os mais importantes que afetam os humanos estão o kuru, a doença de Gerstmann-Sträussler-Scheinker, a síndrome de Creutzfeldt-Jakob e a insônia familiar fatal..
Índice do artigo
Os príons são estruturas de proteínas presentes nas membranas celulares. Essas proteínas têm uma forma ou conformação alterada [PrP (Sc)].
No que se refere à sua multiplicação, ela se dá por meio da conversão de formas, como no caso da doença scrapie. Nesta doença, os príons recrutam PrP (C) (proteínas príon de conformação inalterada) para estimular a conversão para a isoforma PrP (Sc)..
Isso gera uma reação em cadeia que espalha o material infeccioso e, portanto, permite a irrigação da doença. Ainda não se sabe como esse processo de conversão ocorre.
Essas proteínas incomuns, capazes de propagação, não possuem ácidos nucléicos. Prova disso é que são resistentes aos raios X e à radiação ultravioleta. Esses agentes quebram facilmente os ácidos nucléicos.
Proteínas príon, das quais os príons (PrP) são compostos, são encontradas por todo o corpo, não apenas em humanos, mas em outros vertebrados saudáveis. Essas proteínas são geralmente resistentes a proteases (enzimas que catalisam proteínas).
Muito pouco se sabe sobre a utilidade das proteínas príon PrP (C), a forma normal da proteína não infecciosa no corpo humano..
No entanto, alguns pesquisadores conseguiram mostrar que, em camundongos, essas proteínas ativam o reparo da mielina nas células do sistema nervoso periférico. A ausência destes também demonstrou causar desmielinização dessas células nervosas..
O conhecimento que se tem sobre a estrutura dos príons reside principalmente nas investigações realizadas na bactéria. Escherichia coli.
Estudos mostraram que os polipeptídeos de cadeia PrP (C) (normal) e PrP (Sc) (infeccioso) são idênticos na composição de aminoácidos, mas diferem em sua conformação 3D e dobramento..
Esses príons não infecciosos têm 209 aminoácidos em humanos. Eles têm uma ligação dissulfeto. Sua estrutura é alfa-helicoidal, o que significa que possui aminoácidos em forma de espiral (alfa-hélices) e poucas fitas planas de aminoácidos (folhas beta)..
Esta proteína não pode ser separada por centrifugação, o que implica que não é sedimentável. É facilmente digerido pela serina protease de amplo espectro chamada proteinase K.
É uma proteína infecciosa que transforma PrP (C) em isoformas infecciosas de PrP (Sc) e com uma configuração ou forma anormal.
Muito pouco se sabe sobre sua estrutura 3D, porém sabe-se que tem poucas formas helicoidais e mais fios planos ou folhas beta. A mudança para a isoforma é conhecida como o evento principal das doenças por príons.
As proteínas príon celular [Prp (C)] estão localizadas na superfície celular de uma ampla variedade de órgãos e tecidos. Muito pouco se sabe sobre as funções fisiológicas dos príons no corpo. Mesmo assim, experimentos feitos em camundongos indicam possíveis funções, como:
Foi demonstrado que o PrP (C) atua com os receptores de glutamato (ionotrópicos e metabotrópicos). PrP (C) participa como um receptor para oligômeros sinaptotóxicos do peptídeo Aβ de superfície celular.
Em camundongos da família Murinae, descobriu-se que as proteínas príon PrP (C) são expressas poucos dias após a implantação, no desenvolvimento embrionário..
Isso indica que eles desempenham um papel durante o desenvolvimento desses pequenos mamíferos. Papel que segundo os pesquisadores está relacionado à regulação da neuritogênese (produção de axônios e dendritos de neurônios).
Eles também atuam no crescimento axonal. Essas proteínas príon estão até envolvidas no desenvolvimento do circuito cerebelar. Por isso, acredita-se que a ausência desses príons PrP (C) acarreta um atraso no desenvolvimento motor de roedores..
Em estudos sobre a superexpressão de PrP (C) por orientação gênica, verificou-se que a ausência desses príons causa problemas no suprimento de sangue a algumas partes do cérebro (isquemia cerebral aguda).
Isso significa que as proteínas príon funcionam como neuroprotetores. Além disso, foi demonstrado que a superexpressão de PrP (C) pode reduzir ou melhorar as lesões causadas por isquemia..
O papel fisiológico de Prp (C) na manutenção da mielina periférica foi descoberto recentemente.
Durante um estudo de laboratório, foi descoberto que, na ausência da proteína príon, os ratos de laboratório desenvolveram deficiências nos nervos que transportam informações do cérebro e da medula espinhal, no que é chamado de neuropatia periférica..
Existem algumas proteínas que são semelhantes aos príons e estão localizadas em outras partes do corpo que não o cérebro.
A função dessas proteínas é iniciar, regular e / ou controlar a morte celular, quando o organismo está sendo atacado (por virons, por exemplo), evitando a disseminação do patógeno..
Essa função peculiar dessas proteínas faz os pesquisadores pensarem na possível importância dos príons não infecciosos no combate aos patógenos..
Um estudo realizado no Stowers Institute, em Missouri, EUA, mostrou que os príons PrP podem ter um papel na manutenção da memória de longo prazo.
O estudo revelou que certas proteínas príon podem ser controladas para trabalhar na manutenção das funções fisiológicas da memória de longo prazo..
Uma investigação sobre proteínas príon que são expressas em células-tronco do tecido sanguíneo, revelou que todas essas células-tronco (hematopoéticas) expressam proteínas príon em sua membrana celular. Pelo que se acredita que participem do complexo e muito importante processo de renovação celular..
Patologias de origem príon são reconhecidas como distúrbios cerebrais degenerativos progressivos. Eles podem atacar gado, veados, caribus, ovelhas e até mesmo humanos.
Essas doenças são causadas por uma alteração na estrutura das proteínas PrP (C) e cujas funções específicas ainda hoje são incertas. As patologias de príons podem surgir sem uma causa conhecida. Eles podem ter uma origem genética herdada e também podem ser transmitidos de forma infecto-contagiosa.
Os príons causam doenças familiares, esporádicas e contagiosas. As doenças familiares por príons são aquelas que são hereditárias. As patologias esporádicas são as mais comuns e ocorrem sem causas conhecidas..
As doenças contagiosas são consideradas raras, são transmitidas de pessoa para pessoa, de animal para animal, de pessoa para animal e vice-versa. As causas são múltiplas e vão desde o consumo de carne contaminada, canibalismo, transfusões, até a manipulação de equipamento cirúrgico contaminado.
As doenças de príon mais comuns são:
Considerada a doença priônica mais comum entre os seres humanos, é uma doença cosmopolita, ou seja, tem distribuição mundial. Pode ser hereditário (familiar), esporádico ou infeccioso.
Os pacientes apresentam sintomas como demência, espasmos ou movimentos involuntários repentinos e deficiências do sistema nervoso central.
Dependendo do tratamento e da forma da doença, a morte pode ocorrer entre 4 meses a 2 anos após a aquisição da doença. O diagnóstico é difícil de fazer, geralmente é feito post morten, durante a autópsia.
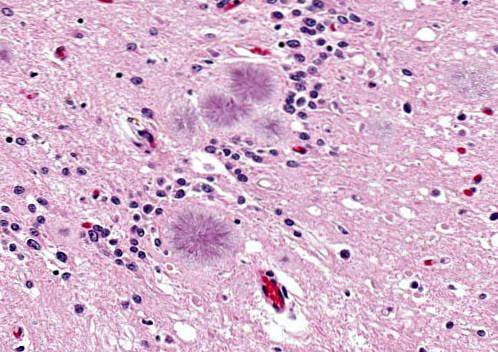
É uma doença causada por príons em um processo infeccioso cerebral hereditário ou autossômico dominante. A doença se manifesta em pessoas de 40 a 60 anos.
Essas pessoas manifestam problemas para articular palavras (disartria), espasmos ou movimentos involuntários repentinos, sendo a agressividade frequente.
Eles se apresentam com degeneração cerebelar acompanhada por uma marcha instável. Também é possível observar hiporreflexia, surdez, paralisia do olhar, demência, entre outros sintomas. A expectativa de vida é de aproximadamente 5 anos ou um pouco mais.
É uma doença muito rara, a ponto de sua taxa de ocorrência ser de 2 a 3 casos por 100 milhões de habitantes. A patologia é semelhante à doença de Gerstmann-Sträussler-Scheinker.
As manifestações clínicas da proteína indicam baixa resistência às proteases, algumas são mais e outras menos sensíveis a essas enzimas..
Os sintomas que os pacientes apresentam são: problemas de fala e deficiência cognitiva, perda de neurônios na área onde o cérebro controla os movimentos e realiza a coordenação muscular.
A doença é frequente em pacientes idosos (70 anos) e o tempo de vida estimado uma vez infectado é de aproximadamente 20 meses.
É uma doença hereditária ou familiar, também pode ocorrer esporadicamente. A doença é conhecida por ser causada por uma mutação hereditária ou autossômica dominante.
Os pacientes apresentam sintomas como problemas cumulativos para dormir e manter o sono, demência, déficit cognitivo, até problemas de hipertensão, taquicardia, hiperidrose e outros..
A idade que afeta é bastante ampla, variando entre 23 e 73 anos, porém a média de idade é de 40 anos. O tempo de vida após a infecção é de pouco mais de 6 anos.
Esta doença por príon só foi detectada nos habitantes de Papua-Nova Guiné. É uma doença relacionada ao canibalismo e à tradição cultural do rito do luto pelos mortos, onde essas pessoas comem cérebro ou carne humana..
Pessoas que são portadoras da doença geralmente apresentam movimentos involuntários e incontroláveis em diferentes partes do corpo.
Apresentam tremores, perda de controle dos movimentos e perda da coordenação muscular. A expectativa de vida das pessoas infectadas é de dois anos.
Entre as patologias produzidas por príons em animais está a encefalopatia espongiforme bovina. Esta doença causou estragos na Europa, na saúde pública, dos animais e na economia dos países afetados.
Outras doenças em animais incluem scrapie, encefalopatia transmissível de vison, doença debilitante crônica (em cervos) e encefalopatia espongiforme felina..
Essas doenças, como as que se apresentam em humanos, carecem de um tratamento eficaz, por isso a prevenção é essencial, principalmente após infecções em humanos que ocorreram em decorrência do consumo de carne de vacas infectadas..
Até o momento, não há cura conhecida para doenças por príons. O tratamento é sintomático. Os pacientes são aconselhados a planejar cuidados paliativos e testes genéticos e aconselhamento para os membros da família são recomendados.
Uma grande variedade de medicamentos foi testada em pacientes com doenças de príon, como antivirais, antitumores, medicamentos para doenças como Parkinson, tratamentos para imunossupressão, antibióticos, antifúngicos e até antidepressivos..
No entanto, atualmente não há evidências que indiquem que alguns deles reduzem os sintomas ou melhoram a sobrevida dos pacientes..
Os príons são resistentes a uma variedade de mudanças físicas e químicas. No entanto, diferentes técnicas são utilizadas para evitar a contaminação de pacientes com instrumentos cirúrgicos contaminados..
Entre as técnicas mais utilizadas está a esterilização do equipamento em autoclave a 132 ° C por uma hora e, a seguir, imersão dos instrumentais em hidróxido de sódio por pelo menos mais uma hora..
Por outro lado, a Organização Mundial da Saúde (OMS) desenvolveu medidas para prevenir a propagação de doenças por príons. Essa organização estabelece normas para o manuseio de tecidos proibidos ou potencialmente perigosos, como: olhos, cérebro, intestino, amígdalas e medula espinhal.



Ainda sem comentários