O Síndrome de Cornelia de Lange é uma patologia de origem genética que se caracteriza pela presença de um atraso cognitivo significativo acompanhado por vários traços físicos malformativos.
No nível clínico, três cursos clínicos diferenciais são observados: grave, moderado e leve. Os sinais e sintomas são geralmente constituídos por uma configuração facial atípica, malformações musculoesqueléticas e atraso no desenvolvimento cognitivo e psicomotor. Além disso, outros tipos de anormalidades relacionadas a malformações cardíacas, pulmonares e / ou digestivas podem ser distinguidas..
Quanto à origem da síndrome Cornelia de Lange, sua etiologia tem sido relacionada à presença de mutações específicas nos genes SMC3, SMC1A, NIPBL, entre outros. O diagnóstico é fundamentalmente clínico, feito com base em características físicas e cognitivas. No entanto, geralmente é acompanhado por um teste genético confirmatório.
O tratamento é voltado para a detecção e tratamento de complicações médicas. Medicina, fonoaudiologia, intervenção neuropsicológica e educação especial são essenciais.
Índice do artigo
Esta síndrome foi inicialmente descrita pela Dra. Cornelia de Lange em 1933. Sua pesquisa foi baseada no estudo de dois pacientes com idade entre 6 e 17 meses. Seu quadro clínico era caracterizado por um grave atraso no crescimento físico e desenvolvimento intelectual associado a vários traços malformativos..
Dada a semelhança dos dois casos, o primeiro relato clínico dessa patologia pressupôs a existência de uma causa etiológica comum e pública..
Anteriormente, Brachmann (1916) havia conseguido publicar alguns dados de autópsia de um paciente em idade infantil com algumas características compatíveis com a síndrome de Cornelia de Lange..
Atualmente, o quadro clínico dessa síndrome foi classificado em três fenótipos diferenciais: grave, moderado e leve..
A síndrome Cornelia de Lange é uma doença genética rara de natureza congênita, ou seja, suas características clínicas são evidentes desde o nascimento. É definida como uma doença multissistêmica com sintomas associados a retardo do desenvolvimento físico e cognitivo, malformações crânio-faciais ou malformações musculoesqueléticas..
Embora o curso clínico e a gravidade desta síndrome possam variar consideravelmente entre os afetados, é uma doença com alta taxa de mortalidade..
Pessoas com síndrome de Cornelia de Lange são caracterizadas por terem uma configuração facial atípica ou característica e um atraso no crescimento / desenvolvimento pré e pós-natal..
O mais comum é que apareçam problemas de aprendizagem, atraso na aquisição da linguagem ou marcha e anormalidades comportamentais.
A síndrome Cornelia de Lange é uma doença rara na população em geral, geralmente classificada como uma doença rara. Os dados epidemiológicos não são exatamente conhecidos. Sua incidência foi estimada em um caso por 10.000-30.000 nascimentos..
Até o momento, podemos encontrar mais de 400 casos diferentes de síndrome de Cornelia de Lange descritos na literatura médica e experimental..
É uma patologia que pode afetar ambos os sexos em igual número. Alguns autores como Gutiérrez Fernández e Pacheco Cumani (2016) sugerem um ligeiro predomínio do sexo feminino, com uma relação de 1,3 / 1.
Em relação aos demais fatores sociodemográficos, a pesquisa atual não identificou uma prevalência diferencial associada a países ou grupos étnicos e / ou raciais específicos..
Boa parte dos casos diagnosticados são esporádicos, embora várias famílias afetadas tenham sido identificadas com um padrão claro de herança dominante..
Os sinais e sintomas da síndrome Cornelia de Lange são caracterizados por seu amplo padrão de envolvimento.
Essa doença é definida pela presença de características faciais, malformações musculoesqueléticas nos membros superiores e inferiores, retardo de crescimento pré e pós-natal generalizado, juntamente com o desenvolvimento de outras anormalidades físicas..
A seguir, descreveremos algumas das características clínicas mais frequentes na síndrome de Cornelia de Lange:
Em mais de 90% das pessoas afetadas pela síndrome de Cornelia Lange, é possível identificar atraso no desenvolvimento físico ou hipocrescimento global. O crescimento é geralmente afetado tanto no período pré-natal quanto no pós-natal.
As características mais comuns em recém-nascidos são:
Essas condições geralmente duram até a idade adulta. Nele pode-se distinguir um tumor localizado abaixo do esperado para o sexo e idade biológica da pessoa afetada.
Junto com esse tipo de alteração, algumas anormalidades relacionadas à alimentação podem ser identificadas. A dificuldade em engolir ou mastigar alimentos é comum durante os primeiros estágios da vida.
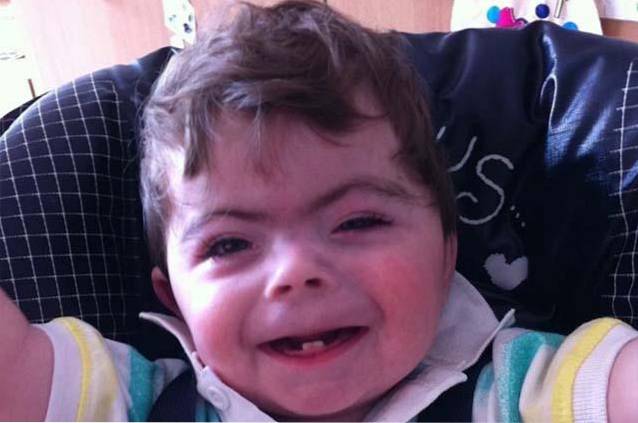
A combinação de alterações cranianas e faciais resulta no desenvolvimento de um fenótipo facial característico em pessoas que sofrem da síndrome de Cornelia de Lange..
Algumas das anomalias mais comuns incluem:
Atraso no desenvolvimento cognitivo e psicomotor constitui um dos achados clínicos centrais na síndrome de Cornelia Lange. Uma lenta aquisição de habilidades relacionadas à atividade motora ou mental é geralmente identificada.
Os marcos mais afetados são a aquisição do sentar, o sorriso afetivo, o balbucio, o movimento independente, a emissão das primeiras palavras, a compreensão e ordens, a alimentação, a deambulação ou o banheiro independente.
Em muitos dos afetados, um QI médio associado a deficiência intelectual leve ou moderada pode ser identificado.
O comportamento das pessoas afetadas pela síndrome de Cornelia de Lange geralmente apresenta algumas características distintas:
A síndrome Cornelia de Lange também está associada ao desenvolvimento de várias complicações médicas.
As causas mais frequentes de morte ou agravamento do estado de saúde das pessoas afetadas estão relacionadas com:
A variabilidade dos sinais e sintomas da síndrome de Cornelia de Lange permitiu uma classificação de seu curso clínico:
Geralmente é o mais sério. As alterações e anomalias são caracterizadas pela presença de vegetação rasteira, malformações musculoesqueléticas, características faciais anormais, limitação da mobilidade articular, atraso cognitivo e outras complicações médicas (auditivas, oculares, digestivas, reno-urológicas, cardíacas e genitais)..
Nesse subtipo, as alterações físicas costumam ser pouco evidentes, principalmente nas extremidades. Os afetados geralmente não apresentam déficits intelectuais graves. O mais comum é que o diagnóstico seja feito além da fase neonatal.
Seu curso clínico é caracterizado fundamentalmente pela variabilidade clínica. As características faciais estão presentes na maioria dos casos, mas a expressão do resto das anomalias é variável..
A origem da síndrome Cornelia Lange está associada à presença de anormalidades genéticas. Nos casos examinados, foi possível identificar mutações específicas em 5 genes diferentes: NIPBL, SMC1A, HDAC8, RAD21 e SMC3.
A alteração mais comum está relacionada ao gene NIPBL, identificado em mais da metade dos afetados. O resto das anomalias genéticas são menos frequentes.
Todos esses genes desempenham papel de destaque na produção de proteínas relacionadas ao complexo coesina, responsáveis pela regulação da estrutura e organização cromossômica, estabilização da informação genética nas células e reparo do DNA..
Além disso, eles também cumprem várias funções fundamentais no desenvolvimento pré-natal das extremidades, da face e de outras regiões e sistemas do corpo..
O diagnóstico da síndrome Cornelia de Lange é clínico. Atualmente não há nenhum teste laboratorial que indique definitivamente sua presença. Na área médica, o mais comum é utilizar os critérios diagnósticos propostos por Kline et al..
Referem-se à identificação de anomalias craniofaciais, em crescimento e desenvolvimento, nas extremidades, alterações neurossensoriais e cutâneas, distúrbios comportamentais, etc..
Além disso, é importante realizar uma análise genética molecular para identificar a presença de mutações associadas à síndrome de Cornelia de Lange..
Embora não haja cura para a síndrome de Cornelia de Lange, sua abordagem terapêutica envolve o desenho de um acompanhamento médico contínuo juntamente com o tratamento das complicações..
Os autores Gil, Ribate e Ramos (2010) apontam algumas das abordagens mais utilizadas..



Ainda sem comentários