
O Síndrome de Joubert é uma doença de origem genética que se caracteriza por uma diminuição do tônus muscular, problemas de coordenação, movimentos oculares anormais, padrões respiratórios alterados e deficiência intelectual (Joubert Syndrome Foundation, 2016).
Todas essas alterações são devidas a uma transmissão genética autossômica que causará importantes anormalidades cerebrais, redução do vermis cerebelar, bem como anormalidades na estrutura do tronco encefálico (National Institute of Neurological Disorders and Stroke, 2016).
Além disso, a síndrome de Joubert faz parte de um grupo de doenças chamadas ciliopatias, que envolvem uma disfunção de uma parte das células denominadas cílios. Joubert Syndrome Foundation, 2016).
A descrição inicial dessa patologia foi feita por Marie Joubert et al., Em 1968, na qual foram descritos quatro casos. Nos pacientes houve ausência parcial ou total do vérmis cerebelar, síndrome da ampnéia-hipernéia episódica neonatal, movimentos oculares anormais, ataxia e retardo mental (Angemi e Zucotti, 2012).
Além disso, essa síndrome também foi associada a diferentes alterações multiorgânicas, como fibrose hepática, polidactilia, nefronoptise ou distrofia retinal (Angemi e Zucotti, 2012).
Em termos de tratamento, atualmente não há cura para a síndrome de Joubert. As intervenções terapêuticas visam o controle e suporte sintomático, a estimulação física e intelectual das crianças e a terapia ocupacional (Instituto Nacional de Doenças Neurológicas e Derrame, 2016).
Índice do artigo
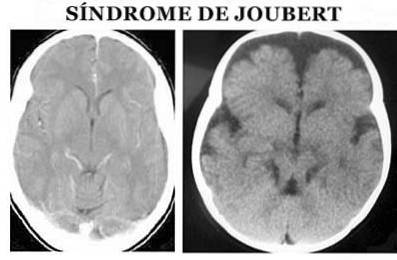
A síndrome de Joubert (JS) é um tipo de patologia de origem genética que se caracteriza por uma malformação congênita nas áreas do tronco encefálico e agenesia (ausência parcial ou completa) ou hipoplasia (desenvolvimento incompleto) do vermis cerebelar, que pode causar (Ophatnet , 2009).
Mais especificamente, a nível anatômico é caracterizado pelo chamado sinal molar do mesencéfalo: agenesia ou hipoplasia do vermis cerebelar, estreitamento dos pedúnculos cerebelares superiores com espessamento, alongamento e falta de decussação e fossa interpeduncular profunda (Angemi e Zuccoti, 2012).
É um distúrbio que pode afetar muitas áreas e órgãos do corpo, portanto, os sinais e sintomas variam consideravelmente entre as pessoas afetadas (U.S. National Library of Medicine, 2011).
A maioria das pessoas afetadas sofre de tônus muscular enfraquecido (hipotonia) e dificuldades de coordenação motora (Ataxia). Outras características são: episódios de respiração alterada, nistagmo (movimento involuntário e arrítmico dos olhos), atraso no desenvolvimento motor e dificuldades intelectuais variáveis (U.S. National Library of Medicine, 2011).
A prevalência da síndrome de Joubert foi estimada em aproximadamente 1 / 80.000 a 1 / 100.000.000 nascidos vivos. Em todo o mundo, mais de 200 casos clínicos foram registrados (Angemi e Zuccoti, 2012).
Muitos especialistas consideram esses números subestimados, uma vez que a síndrome de Joubert tem uma ampla gama de afetações e é amplamente subdiagnosticada (U.S. National Library of Medicine, 2011).
Muitos dos sintomas clínicos da síndrome de Joubert são mais do que evidentes na infância, muitas crianças afetadas apresentam atrasos motores significativos (National Organization for Rare Disease, 2011).
As características mais comuns do curso clínico são: falta de controle muscular (ataxia), padrões respiratórios alterados (hipercapnia), apnéia do sono, movimentos oculares anormais (nistagmo) e baixo tônus muscular (National Organization for Rare Disease, 2011).
Por outro lado, algumas das alterações que podem estar associadas à síndrome de Joubert incluem: desenvolvimento alterado da retina, anormalidades na íris, estrabismo, alterações renais e / ou hepáticas, protrusão das membranas que revestem o cérebro, entre outras ( Organização Nacional para Doenças Raras, 2011).
Todas as alterações derivadas desta síndrome são englobadas em várias áreas: alterações neurológicas, oculares, renais e musculoesqueléticas (Bracanti et al., 2010).
As alterações neurológicas mais características da síndrome de Joubert são Bracanti et al., 2010): hipotonia, ataxia, atraso generalizado no desenvolvimento, alterações intelectuais, alteração dos padrões respiratórios e movimentos oculares anormais.
No nível físico, a retina é um dos órgãos afetados pela síndrome de Joubert. As alterações neste órgão aparecem na forma de distrofia retiniana, devido a uma degeneração progressiva das células responsáveis pela foto recepção..
Clinicamente, as alterações oculares podem variar de cegueira retiniana congênita a degeneração retiniana progressiva.
Por outro lado, também é possível observar a presença do coloboma. Esta alteração ocular é um defeito congênito que afeta a íris ocular e aparece como um orifício ou fenda.
Patologias relacionadas à função renal afetam mais de 25% das pessoas afetadas pela síndrome de Joubert.
Em muitos casos, as anormalidades renais podem permanecer assintomáticas por vários anos ou começar a se manifestar com sinais inespecíficos, até se apresentarem como insuficiência renal aguda ou crônica..
Desde as primeiras descrições desta patologia, um achado clínico frequente é a polidactialia (uma doença genética que aumenta o número de dedos das mãos ou dos pés).
Além disso, também é comum observar anomalias orofaciais ou estruturais ao nível da coluna..
Estudos experimentais classificaram a síndrome de Joubert como um transtorno autossômico recessivo (National Organization for Rare Disease, 2011).
Um distúrbio genético autossômico recessivo significa que duas cópias de um gene anormal devem estar presentes para que o traço ou doença apresente (National Institutes of Health, 2014).
Portanto, uma alteração genética recessiva ocorre quando uma pessoa herda o mesmo gene anormal para a mesma característica de cada um dos pais. Se um indivíduo receber apenas uma cópia do gene relacionado à doença, será portador, mas não apresentará sintomas (National Organization for Rare Disease, 2011).
Além disso, pelo menos dez genes foram identificados como uma das possíveis causas da síndrome de Joubert (National Organization for Rare Disease, 2011).
Uma mutação no gene AHI1 é responsável por essa condição patológica em aproximadamente 11% das famílias afetadas. Em pessoas com esta alteração genética, as alterações da visão são comuns devido ao desenvolvimento de distrofia retiniana (National Organization for Rare Disease, 2011).
A mutação do gene nphp1 é a causa de aproximadamente 1-2% dos casos de síndrome de Joubert. Em indivíduos com esta alteração genética, as alterações renais são comuns (National Organization for Rare Disease, 2011).
Por outro lado, uma mutação do gene CEP290 é a causa de 4-10% dos casos de síndrome de Joubert (National Organization for Rare Disease, 2011).
Além disso, mutações nos genes TME67, JBTS1, JBTS2, JBTS7, JBTS8 e JBTS9 também estão relacionadas ao desenvolvimento da síndrome de Joubert (National Organization for Rare Disease, 2011).
O diagnóstico da síndrome de Joubert é feito com base nos sintomas físicos. É necessária a realização de um exame físico detalhado, bem como a utilização de diversos testes diagnósticos, principalmente imagens de ressonância magnética (Ophatnet, 2009)..
Além disso, os testes genéticos moleculares também são frequentemente usados para identificar alterações genéticas que foram demonstradas em 40% dos casos de síndrome de Joubert (National Organization for Rare Disease, 2011).
Por outro lado, também é possível fazer o diagnóstico pré-natal dessa patologia por meio da ultrassonografia fetal e da análise molecular, principalmente em famílias com história genética de síndrome de Joubert (Ophatnet, 2009)..
Quando as características mais características da síndrome de Joubert ocorrem em combinação com uma ou mais patologias físicas adicionais, um diagnóstico de síndrome de Joubert e distúrbios relacionados (JSRD) pode ser feito (U.S. National Library of Medicine, 2011).
Portanto, dependendo do tipo de patologia relacionada à presença da síndrome de Joubert, podemos encontrar subtipos desta. No entanto, o sistema de classificação da síndrome de Joubert ainda está em fase de evolução devido à descoberta de contribuições genéticas e ao maior conhecimento das correlações fenotípicas..
Podemos, portanto, encontrar (Bracanti et al., 2010):
O tratamento usado na síndrome de Joubert é sintomático e de suporte das patologias subjacentes. Além de intervenções farmacológicas, é comum o uso de estimulação física e cognitiva precoce (National Institute of Neurological Disorders and Stoke, 2016).
Quando as alterações respiratórias são significativas, principalmente nas fases iniciais da vida, é necessário monitorar a função respiratória (National Institute of Neurological Disorders and Stoke, 2016).
Por outro lado, a identificação e controle da degeneração ocular, complicações renais e o resto das complicações relacionadas à síndrome de Joubert, devem ser realizados o mais cedo possível para ajustar as medidas terapêuticas (National Institute of Neurological Disorders and Stoke, 2016 ).


Ainda sem comentários