
O Síndrome de MELAS É um tipo de doença mitocondrial de origem hereditária que se caracteriza pelos distúrbios neurológicos que causa. Essa patologia é fundamentalmente definida pela apresentação de encefalopatia mitocondrial, acidose láctica e episódios semelhantes a AVC..
A nível clínico, os sinais e sintomas da síndrome MELAS costumam ser evidentes antes dos 40 anos e estão relacionados com o sofrimento de convulsões, distúrbios de consciência ou acidentes vasculares cerebrais, entre outros..
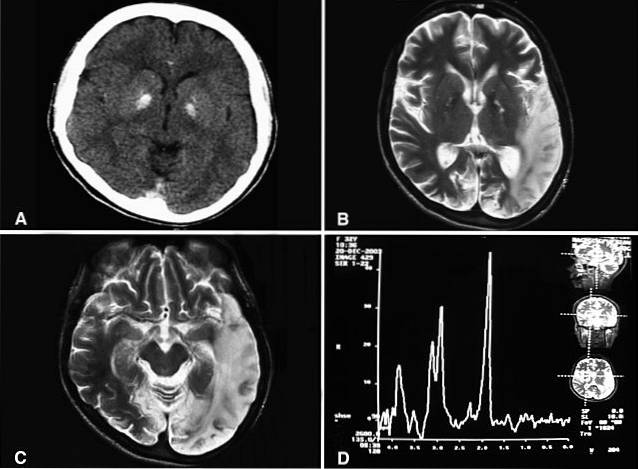
Essa patologia tem origem etiológica genética associada a mutações específicas no DNA mitocondrial e anormalidades nas cadeias enzimáticas. Em caso de suspeita clínica, o diagnóstico da síndrome MELAS geralmente inclui vários exames laboratoriais, como eletroencefalografia (EEG), tomografia axial computadorizada (TC) craniana, ressonância magnética (MRI) e estudo genético.
Não há cura para a síndrome MELAS. As abordagens terapêuticas enfocam o controle dos sintomas e os cuidados paliativos. Dada a natureza degenerativa e crônica da doença MELAS, o prognóstico médico está associado a complicações importantes (distúrbios cardiopulmonares, renais, metabólicos e neurológicos).
Índice do artigo
A síndrome MELAS foi inicialmente descrita por Shapiro e seu grupo de trabalho em 1975. No entanto, foi Pavlakis (1984) que usou o nome MELAS como um acrônimo para suas manifestações mais características..
Em seu relatório clínico, Pavlakis referiu-se a um curso clínico caracterizado por uma combinação de convulsões, comprometimento progressivo da linguagem, acidose láctica e rompimento das fibras musculares vermelhas..
Foram Pavlakis e Hirado que estabeleceram os critérios clínicos para a síndrome MELAS: convulsões, demência, acidose láctica, fibras vermelhas irregulares e episódios semelhantes a derrames antes dos 40 anos de idade..
A apresentação dessa síndrome é amplamente variável e seu curso clínico geralmente é evidente antes da quarta década de vida. O prognóstico médico geralmente é ruim, os afetados progridem com complicações médicas importantes até a morte.
A síndrome MELAS é uma doença rara que geralmente começa na infância ou adolescência, geralmente entre 2 e 15 anos de idade. Afeta especialmente o sistema nervoso e a estrutura muscular do corpo.
Algumas de suas características clínicas incluem convulsões, dores de cabeça recorrentes, vômitos, perda de apetite, episódios semelhantes a derrames, alteração da consciência, alterações de visão e audição e outros tipos de anormalidades motoras e cognitivas..
Esta síndrome deve seu nome às características clínicas cardinais que a definem: encefalomiopatia mitocondrial (encefalomopatia mitocondrial) eu; acidose láctica (acidose láctica) A; episódios semelhantes a derrames S (genética).
A síndrome MELAS é frequentemente categorizada como uma doença mitocondrial ou encefalomiopatia mitocondrial.
As doenças mitocondriais constituem um amplo grupo de patologias caracterizadas pela presença de alterações neurológicas de origem hereditária causadas por mutações específicas no DNA nuclear ou mitocondrial..
A mitocôndria é um tipo de organela celular localizada no citoplasma. Esta organela é essencial para o metabolismo energético das células do nosso corpo. É responsável pela obtenção de energia de um processo oxidativo para a produção de ATP. Além disso, esse componente tem sua própria dotação genética, o DNA mitocondrial..
O processo de produção de energia envolve uma ampla variedade de mecanismos bioquímicos, sendo a anomalia comum nas doenças mitocondriais a alteração da fase final do mecanismo oxidativo..
Esta é a cadeia respiratória mitocondrial que resulta em uma diminuição significativa na produção de energia em sua forma ATP. Devido a isso, as doenças mitocondriais podem se apresentar com importantes anormalidades multissistêmicas, incluindo alterações neurológicas e cerebrovasculares.
Os mais comuns são a síndrome MERRF, a síndrome de Kearns-Sayre e a síndrome MELAS..
A síndrome MELAS é uma doença rara na população em geral. Embora sua prevalência específica não seja conhecida com precisão, é uma das doenças mais comuns classificadas dentro das doenças mitocondriais..
Globalmente, as doenças mitocondriais têm uma prevalência de aproximadamente 1 caso em 4.000 pessoas em todo o mundo.
No que diz respeito às características sociodemográficas, a nível internacional não foi identificada predileção por sexo, grupo étnico / racial ou origem geográfica particular..
A síndrome MELAS é definida pela presença de três achados clínicos cardinais: encefalopatia mitocondrial, acidose láctica e episódios semelhantes a AVC..
Encefalopatia é o termo normalmente utilizado para designar aquelas doenças ou patologias cujo curso clínico heterogêneo tem sua origem em anormalidades estruturais e funcionais do sistema nervoso central..
No nível neurológico, a síndrome MELAS é caracterizada pela apresentação de crises recorrentes. As convulsões são definidas pelo desenvolvimento de episódios temporários de agitação motora excessiva, presença de movimentos musculares bruscos e involuntários, percepção de sensações anormais ou alteração da consciência.
As convulsões podem ter curso diferencial, sendo focais ou generalizadas:
A gravidade clínica das crises está em sua capacidade potencial de danificar permanentemente as estruturas nervosas, levando a sequelas cognitivas e psicomotoras.
Devido a anormalidades nos mecanismos oxidativos envolvidos na produção de energia no corpo, a síndrome MELAS geralmente envolve um acúmulo anormal e patológico de ácido láctico.
O ácido láctico é uma substância bioquímica resultante da degradação dos carboidratos quando os usamos como forma de energia na presença de baixos níveis de oxigênio (insuficiência respiratória, exercício físico, etc.).
Esta substância é geralmente gerada principalmente nos glóbulos vermelhos e nas células musculares. Em condições normais, o ácido láctico é eliminado do corpo através do fígado. No entanto, a presença de níveis anormalmente elevados leva ao desenvolvimento de acidose..
A acidose geralmente gera anormalidades médicas de grande importância, que podem levar à morte da pessoa afetada.
Alguns dos sintomas característicos dessa condição são náuseas, vômitos, diarréia, letargia, dor gástrica, alteração severa do nível de consciência, anormalidades respiratórias, hipotensão arterial, desidratação e até choque médico..
Episódios semelhantes a AVC são caracterizados por serem semelhantes a sofrer um acidente vascular cerebral ou acidente vascular cerebral. Esses eventos são caracterizados pela presença de alterações neurológicas focais, de aparecimento espontâneo e de duração limitada..
Tendem a afetar preferencialmente as áreas occipitais, gerando distúrbios visuais. No entanto, também suas frequentes anormalidades linguísticas, sensoriais ou motoras.
A identificação de múltiplos processos de multi-infarto em diferentes regiões cerebrais acarreta o sofrimento de uma deterioração cognitiva progressiva, com tendência à demência..
A presença das características clínicas descritas acima leva ao desenvolvimento de vários sinais e sintomas secundários. Embora o curso clínico da síndrome MELAS possa ser muito heterogêneo, o mais comum é observar algumas das seguintes características:
Além desses achados, as manifestações psiquiátricas também são frequentemente comuns na síndrome MELAS. Alguns dos mais comuns incluem:
Em outros casos, outras condições podem ser distinguidas, como:
A síndrome MELAS se deve à presença de alterações no DNA mitocondrial. Esses tipos de anomalias são herdadas dos pais maternos, pois esse tipo de DNA, no caso do pai, é perdido durante a fertilização..
No nível genético, a origem da síndrome MELAS tem sido associada a mutações específicas em vários genes: MT-TV, MT-TL1, MT-TH, MT-ND5, MT-ND1. Este conjunto de genes geralmente está localizado no material genético (DNA) da mitocôndria celular.
Muitos desses genes desempenham um papel essencial na produção de proteínas envolvidas na conversão de açúcares, gorduras e oxigênio em energia. No entanto, outros medeiam a produção de moléculas de tRNA essenciais na construção da estrutura dos aminoácidos..
No diagnóstico da síndrome MELAS, é fundamental identificar um alto índice de suspeita clínica, ou seja, é necessário avaliar todas as características clínicas da pessoa afetada. Em qualquer caso, o exame da história médica individual e materna é altamente relevante..
Para confirmar o diagnóstico e descartar outras patologias, é necessário realizar diversos exames complementares:
Atualmente não há cura para a síndrome MELAS..
O uso de procedimentos experimentais (administração de ácido fólico, tiamina, vitamina C, Coenzima Q10, corticosteróides, etc.) também não conseguiu interromper o progresso desta patologia..
O mais comum é o uso de abordagens médicas com foco no controle dos sintomas e cuidados paliativos.
O manejo dos sinais e sintomas por uma equipe médica multidisciplinar é fundamental: oftalmologistas, nefrologistas, endocrinologistas, neurologistas, cardiologistas, etc..
A síndrome MELAS geralmente tem um curso definido por recorrência, remissão ou apresentação de crises agudas, tornando difícil avaliar com precisão a eficácia de novas abordagens terapêuticas.
Os pacientes afetados inevitavelmente desenvolvem deficiência cognitiva, distúrbios psicomotores, perda de visão e audição e outras complicações médicas até a morte.


Ainda sem comentários