
O Síndrome de Prader-Willi (SPW) é uma patologia multissistêmica de origem genética congênita. É uma doença complexa que afeta o apetite, o crescimento, o metabolismo, o comportamento e / ou a função cognitiva.
A nível clínico, na fase da infância, esta doença é caracterizada pela presença de vários achados médicos, como fraqueza muscular, distúrbios alimentares ou atraso generalizado no desenvolvimento..
Além disso, no nível cognitivo e comportamental, boa parte dos indivíduos afetados pela síndrome de Prader-Willi apresentam deficiência intelectual moderada ou atraso que é acompanhado por vários problemas de aprendizagem e comportamento.
Embora a síndrome de Prader-Willi seja considerada uma doença rara ou incomum, vários estudos indicam que é uma das patologias mais frequentes na área genética. O diagnóstico da doença é feito principalmente com base em achados clínicos e testes genéticos complementares..
Em relação ao tratamento, ainda não foi identificada a cura para a síndrome de Prader-Willi, por isso a abordagem terapêutica é orientada para o tratamento dos sintomas e complicações, sendo a obesidade o achado médico que representa a maior ameaça aos afetados.
Assim, em relação ao prognóstico e qualidade de vida, ambos dependerão da gravidade dos problemas médicos associados e dos distúrbios comportamentais ou cognitivos que podem se desenvolver..
Índice do artigo
Diversos relatos clínicos indicam que a síndrome de Prader-Willi (PWS) foi inicialmente descrita por J. L. Down, em 1887, após o diagnóstico de “polissarcia” em um de seus pacientes..
No entanto, foram os Drs Prader, Labhart e Willi que, em 1956, descreveram outros 9 casos e deram o nome a esta patologia. Além disso, as características e critérios diagnósticos da síndrome de Prader-Willi foram sistematizados por Holm et al..
A síndrome de Prader-Willi é uma alteração genética congênita, ou seja, é uma patologia que está presente desde o momento do nascimento e afetará o indivíduo por toda a vida se não houver intervenção terapêutica curativa..
Esta patologia apresenta curso clínico complexo, caracterizado por inúmeras manifestações médicas.
Embora hoje o fenótipo da síndrome de Prader-Willi seja conhecido de forma mais precisa, isso ocorre nos últimos 25 anos, quando houve avanços significativos na análise e compreensão desta doença..
A expressão da síndrome de Prader-Willis é diversa, tende a afetar múltiplos sistemas e estruturas, sendo a maioria das alterações relacionadas à disfunção hipotalâmica.
O hipotálamo é uma estrutura neurológica que desempenha um papel essencial no controle das funções homeostáticas: regulação da fome, sede, ciclos sono-vigília ou regulação da temperatura corporal..
Além disso, o hipotálamo libera diferentes hormônios para várias glândulas: crescimento, sexual, tireóide, etc..
Por fim, devemos destacar que a síndrome de Prader-Willis também pode aparecer referenciada na literatura médica e experimental com outros termos, como síndrome de Prader-Labhart-Willi ou com a sigla PWS..
Além disso, outros sinônimos são síndrome de Labhart Willi, síndrome de Praser Labhart Willi Fancone ou síndrome de distrofia hipogenital..
A síndrome de Prader-Willi (PWS) é uma doença genética rara. O termo doença rara (ER) é usado para se referir às patologias que são raras ou poucas pessoas que sofrem com isso.
Atualmente, estima-se que a síndrome de Prader-Willi é uma patologia com uma frequência aproximada de 1 caso por 10.000-30.000 pessoas em todo o mundo.
Por outro lado, quanto à distribuição por sexo, observou-se que essa patologia atinge igualmente homens e mulheres, não estando associada a etnias ou regiões geográficas..
Além disso, a síndrome de Prader-Willi é considerada a principal causa de obesidade de origem genética.
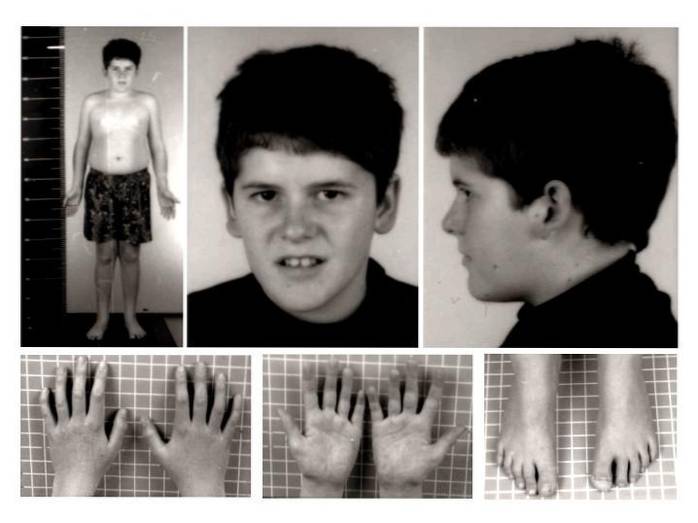
A nível clínico, a síndrome de Prader-Willi tem sido tradicionalmente associada a hipotonia neonatal, hipogonadismo, hiperfagia, obesidade, baixa estatura, atraso generalizado no desenvolvimento, deficiência intelectual moderada, aparência facial atípica e diferentes alterações comportamentais..
Apesar disso, a expressão clínica dessa patologia é muito heterogênea e varia significativamente entre os indivíduos afetados..
Além disso, os sinais e sintomas característicos da síndrome de Prader-Willi tendem a variar com o desenvolvimento biológico, portanto, podemos observar diferentes achados clínicos no período fetal e neonatal, no período da primeira infância ou na primeira infância, na fase escolar e por fim, na fase adolescente..
De forma sistemática, José A. del Barrio del Campo e colaboradores descrevem detalhadamente as alterações mais características na área biomédica, psicomotora, cognitiva e comportamental:
Os sinais e sintomas físicos mais característicos incluem distúrbios como; hipotonia, malformações ou deformidades musculoesqueléticas, peso e altura reduzidos ou baixos, excesso de apetite, obesidade, hipogonadismo, distúrbios do sono, distúrbios respiratórios, traços fáceis atípicos, alteração na regulação da temperatura corporal, entre outros.
Presença ou desenvolvimento de tônus muscular reduzido. A flacidez muscular nesta patologia é especialmente acentuada no pescoço e no tronco, principalmente na fase neonatal e nos primeiros meses de vida. Assim, com o desenvolvimento biológico, o tônus muscular tende a melhorar.
Nesse caso, é comum observar o desenvolvimento de escoliose ou desvio da coluna, mau alinhamento dos membros inferiores (genu valgo) ou a presença de pés planos..
Além disso, outros tipos de anomalias congênitas também podem ser observados, como redução do tamanho dos pés e das mãos, displasia do quadril, presença de seis dedos, entre outros..
Principalmente no momento do nascimento, tanto a altura quanto o peso da criança afetada são menores do que o esperado para seu desenvolvimento e sexo. Apesar do fato de que os valores padrão podem ser alcançados na idade adulta, a taxa de crescimento lento tende a alterar os valores adultos de altura e peso..
É comum observar em pessoas com síndrome de Prader-Willi um apetite insaciável, caracterizado por uma obsessão ou fixação pela comida. Devido à ingestão de grandes quantidades de alimentos, as pessoas afetadas tendem a desenvolver obesidade e outras complicações médicas associadas, como diabetes mellitus tipo II.
A presença de alterações genitais também é frequente. Especificamente, o hipogonadismo ou desenvolvimento parcial da genitália externa é muito comum. Na maioria dos casos, o desenvolvimento puberal não atinge os estágios finais ou adultos.
Ronco, aumento da frequência ou parada respiratória freqüentemente aparecem de forma recorrente durante as fases do sono. Assim, os acometidos tendem a apresentar diversas alterações relacionadas à fragmentação, atraso do sono ou presença de despertares periódicos.
Anormalidades e malformações musculoesqueléticas também podem afetar as características craniofaciais. É possível observar um crânio estreito, estrabismo ocular, pele e cabelos mal pigmentados, boca pequena e lábios finos, malformações dentárias, etc..
Pessoas afetadas pela síndrome de Prader-Willi geralmente têm problemas relacionados à regulação da temperatura corporal, e outro achado significativo é a alta resistência à dor.
Devido à presença de malformações musculoesqueléticas e redução do tônus muscular, o desenvolvimento psicomotor será mais lento, afetando todas as áreas.
Os afetados costumam apresentar graves dificuldades para realizar qualquer tipo de atividade que requeira uma ou mais execuções motoras..
Em relação às limitações cognitivas, a maioria das pessoas afetadas tem deficiência intelectual leve ou moderada.
Além disso, costumam apresentar algumas áreas específicas mais afetadas como processamento sequencial de informações, memória recente ou de curto prazo, resolução de problemas aritméticos, processamento auditivo de informações verbais, alteração da atenção e concentração e presença de rigidez cognitiva..
Por outro lado, a linguagem é outra área que é significativamente afetada em indivíduos com síndrome de Prader-Willi. Atrasos na aquisição de habilidades fonológicas, vocabulário deficiente, alteração da construção gramatical, entre outros, costumam ser observados..
Problemas e alterações comportamentais são outro dos achados típicos que podem ser observados na síndrome de Prader-Willi, normalmente variam dependendo da idade ou estágio maturacional em que a pessoa afetada se encontra, no entanto, alguns dos traços comportamentais mais comuns são:
Diversas investigações atuais têm apontado que as alterações comportamentais tendem a aumentar com a idade e, portanto, tendem a se agravar, afetando as áreas social, familiar e emocional de forma generalizada..
Como apontamos em várias seções acima, a síndrome de Prader-Willi tem origem genética.
Embora atualmente exista uma grande controvérsia sobre os genes específicos responsáveis por esta patologia, todos os dados mostram que a alteração etiológica está localizada no cromossomo 15..
Ao longo do estudo genético desta patologia, houve várias contribuições. Burtler e Palmer (1838) detectaram a presença de anormalidades no braço longo do cromossomo 15 do pai paterno, enquanto Nicholls (1989) observou que em outros casos o distúrbio estava relacionado a alterações cromossômicas da mãe (Rosell-Raga, 2003).
Além disso, a teoria mais aceita sobre a origem dessa patologia é a perda ou inativação de vários genes de expressão paterna que estão localizados na região 15q11-13 do cromossomo 15.
O diagnóstico da síndrome de Prader-Willi tem dois componentes básicos: a análise dos achados clínicos e o teste genético..
No que se refere à detecção de sinais e sintomas indicadores, tanto em bebês quanto em crianças maiores, será imprescindível a realização de um histórico médico detalhado, individual e familiar. Da mesma forma, também é imprescindível a realização de um exame físico e neurológico..
Se com base nesses procedimentos houver suspeita diagnóstica, será necessário prescrever vários exames complementares para determinar a presença de alterações e anormalidades genéticas..
Especificamente, cerca de 90% dos casos são definitivamente diagnosticados por meio de testes de metilação do DNA e outros testes adicionais..
Além disso, também é possível fazer o diagnóstico pré-natal dessa condição médica, principalmente em famílias com história prévia de síndrome de Prader-Willi..
Especificamente, o teste de amniocentese permite a extração de amostras de embriões para a realização dos testes genéticos pertinentes..
Atualmente, não há cura para a síndrome de Prader-Willi. Como em outras doenças raras, os tratamentos se limitam ao controle dos sintomas e à melhoria da qualidade de vida das pessoas afetadas.
Porém, um dos aspectos fundamentais será o controle nutricional e dietético, visto que a obesidade é a principal causa de morbimortalidade nessa patologia..
Por outro lado, a presença de alterações cognitivas e comportamentais exigirá a intervenção de profissionais especializados tanto na reabilitação cognitiva quanto no manejo do transtorno de conduta..


Ainda sem comentários